
Analysis of different colonic transport dynamics in patients with functional constipation based on 16S sequencing technology differential study of intestinal flora in patients with functional constipation
-
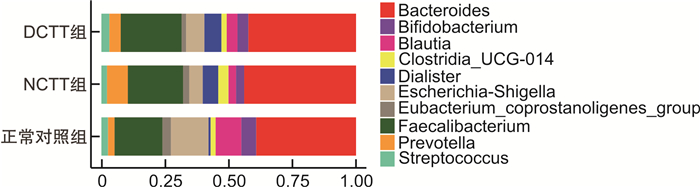
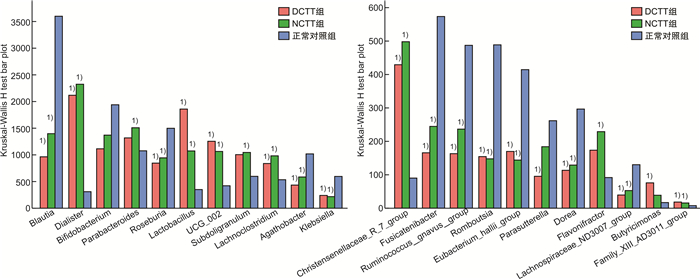
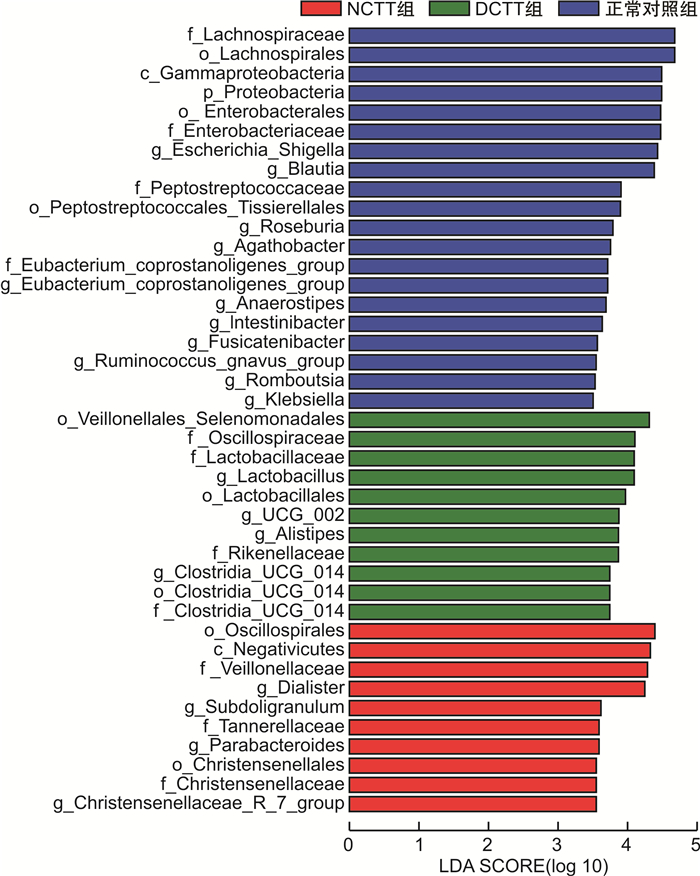
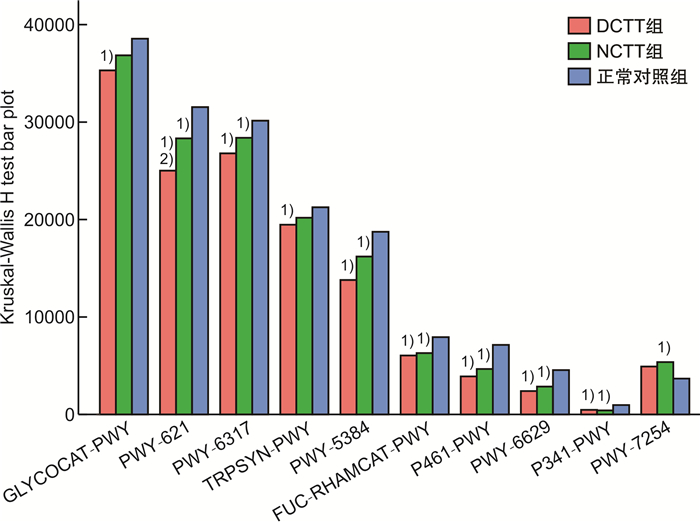
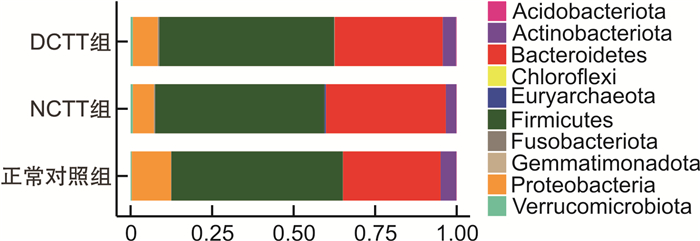
摘要: 目的 运用16S rDNA测序技术分析不同结肠传输动力功能性便秘(FC)患者与正常对照组肠道菌群的差异。方法 招募福建中医药大学附属第二人民医院60例FC患者,根据结肠传输试验结果分为结肠转运时间延迟(DCTT)组20例、结肠转运时间正常(NCTT)组40例,同时招募30名健康志愿者为正常对照组,留取各组新鲜粪便,采用16S rDNA测序技术分析肠道菌群的差异,并对测序结果进行基因功能预测。结果 ①α多态性分析:在菌群丰度指数(Chao1、observed_otus)上DCTT组较NCTT组与正常对照组升高(P < 0.05)。3组多样性指数(shannon、simpson)比较差异无统计学意义(P>0.05)。②3组群落组成分析:3组均以厚壁菌门、拟杆菌门、变形菌门为优势菌门,以拟杆菌属、粪杆菌属为优势菌属。在属水平上,Blautia、Roseburia、Agathobacter、Klebsiella、Fusicatenibacter、Ruminococcus_gnavus_group、Romboutsia、Eubacterium_hallii_group、Dorea、Lachnospiraceae_ND3007_group菌属DCTT组与NCTT组较正常对照组水平下降(P < 0.05),但DCTT组与NCTT组差异无统计学意义(P>0.05),Dialister、Lactobacillus、UCG_002、Lachnoclostridium、Christensenellaceae_R_7_group、Family_XⅢ_AD3011_group菌属DCTT组与NCTT组较正常对照组升高(P < 0.05),但DCTT组与NCTT组比较差异无统计学意义(P>0.05);Parabacteroides、Subdoligranulum、Flavonifractor菌属NCTT组较正常对照组升高(P < 0.05),与DCTT组比较差异无统计学意义(P>0.05),Butyricimonas菌属DCTT组较正常对照组升高(P < 0.05),与NCTT组比较差异无统计学意义(P>0.05),Parasutterella菌属DCTT组较正常对照组下降(P < 0.05),与NCTT组比较差异无统计学意义(P>0.05)。③LEfSe分析显示正常对照组以Blautia、Roseburia、Agathobacter、Escherichia-Shigella、Eubacterium coprostanoligenes group、Anaerostipes、Intestinimonas、Fusicatenibacter、Ruminococcus_gnavus_group、Romboutsia、Klebsiella为生物学标记,DCTT以Lactobacillus、UCG_002、Alistipes、Clostridia_UCG_014为生物学标记,NCTT以Dialister、Subdoligranulum、Parabacteroides、Christensenellaceae_R_7_group生物学标记。④功能预测分析:LYCOCAT-PWY、TRPSYN-PWY DCTT组较正常对照组下降(P < 0.05),NCTT组与正常对照组比较差异无统计学意义(P>0.05);PWY-621 DCTT组与NCTT组均较正常对照组下降(P < 0.05);PWY-6317、PWY-5384、FUC-RHAMCAT-PWY、P461-PWY、PWY-6629、P341-PWY DCTT组与NCTT组均较正常对照组下降(P < 0.05);PWY-7254 NCTT组较正常对照组升高(P < 0.05),DCTT组与正常对照组比较差异无统计学意义(P>0.05),提示FC患者肠道菌群可能与短链脂肪酸及色氨酸代谢关系密切。结论 不同结肠传输动力的FC患者存在不同肠道菌群特征,FC不同结肠传输动力的特征性菌群异常可能是FC肠道动力障碍的重要因素。Abstract: Objective To analyze the differences in intestinal flora between patients with functional constipation(FC) of different colonic transit times and a normal group using 16S rDNA sequencing technology.Methods Sixty FC patients from the Second People's Hospital affiliated with Fujian University of Traditional Chinese Medicine were recruited and divided into two groups based on the results of colonic transit tests: 20 cases in the delayed colonic transit time(DCTT) group and 40 cases in the normal colonic transit time(NCTT) group. Additionally, 30 healthy volunteers were recruited as a normal control group. Fresh feces were collected from each group, and 16S rDNA sequencing was used to analyze differences in gut microbiota. The sequencing results were subjected to gene function prediction.Results (1) α diversity analysis: The abundance indices Chao1 and observed_otus in the DCTT group were higher than those in the NCTT group and the normal control group(P < 0.05). There were no statistically significant differences in diversity(Shannon index, Simpson index) among the three groups(all P>0.05). (2) Community Composition Analysis: The predominant phyla in all three groups were Firmicutes, Bacteroidetes, and Proteobacteria, with Bacteroides and Faecalibacterium being the dominant genera. At the genus level, Blautia, Roseburia, Agathobacter, Klebsiella, Fusicatenibacter, Ruminococcus_gnavus_group, Romboutsia, Eubacterium_hallii_group, Dorea, and Lachnospiraceae_ND3007_group were decreased in both the DCTT and NCTT groups compared to the normal control group(P < 0.05), but there were no significant differences between the DCTT and NCTT groups(P>0.05). Genera such as Dialister, Lactobacillus, UCG_002, Lachnoclostridium, Christensenellaceae_R_7_group, and Family_XⅢ_AD3011_group were increased in the DCTT and NCTT groups compared to the normal control group(P < 0.05), with no significant differences between the DCTT and NCTT groups(P>0.05). Genera such as Parabacteroides, Subdoligranulum, and Flavonifractor were increased in the NCTT group compared to the normal control group(P < 0.05), with no significant differences compared to the DCTT group(P>0.05). Butyricimonas were increased in the DCTT group compared to the normal control group(P < 0.05), with no significant differences compared to the NCTT group(P>0.05). Parasutterella was decreased in the DCTT group compared to the normal control group(P < 0.05), with no significant differences compared to the NCTT group(P>0.05). (3) LEfSe Analysis: The normal control group had Blautia, Roseburia, Agathobacter, Escherichia-Shigella, Eubacterium coprostanoligenes group, Anaerostipes, Intestinimonas, Fusicatenibacter, Ruminococcus_gnavus_group, Romboutsia, and Klebsiella as biomarkers. The DCTT group had Lactobacillus, UCG_002, Alistipes, and Clostridia_UCG_014 as biomarkers. The NCTT group had Dialister, Subdoligranulum, Parabacteroides, and Christensenellaceae_R_7_group as biomarkers. (4) Functional Prediction Analysis: The LYCOCAT-PWY and TRPSYN-PWY pathways were decreased in the DCTT group compared to the normal control group(P < 0.05), with no significant differences between the NCTT group and the normal control group(P>0.05). The PWY-621 pathway was decreased in both the DCTT and NCTT groups compared to the normal control group(P < 0.05). Pathways such as PWY-6317, PWY-5384, FUC-RHAMCAT-PWY, P461-PWY, PWY-6629, and P341-PWY were decreased in both the DCTT and NCTT groups compared to the normal control group(P < 0.05). The PWY-7254 pathway was increased in the NCTT group compared to the normal control group(P < 0.05), with no significant differences between the DCTT group and the normal control group(P>0.05), suggesting that gut microbiota in FC patients may be closely related to short-chain fatty acid and tryptophan metabolism.Conclusion Patients with FC of different colonic transit dynamics exhibit distinct gut microbiota characteristics. The characteristic dysbiosis of gut microbiota in FC patients with different colonic transit dynamics may be an important factor in the colonic motility disorders observed in FC.
-
-
表 1 FC患者与正常对照组肠道菌群丰度与多样性分析
M(P25,P75) 组别 例数 丰度 多样性 Chao1 observed_otus shannon simpson DCTT组 20 520.47(451.48,606.10)1) 515.00(445.50,599.00)1) 6.87(6.65,7.27) 0.981(0.970,0.985) NCTT组 40 466.34(373.17,539.99) 454.50(367.00,531.50) 6.80(6.25,7.07) 0.980(0.967,0.984) 正常对照组 30 435.65(387.98,468.36) 431.50(385.25,459.00) 6.68(6.50,6.89) 0.976(0.964,0.981) 与正常对照组比较,1)P < 0.05。  下载: 导出CSV
下载: 导出CSV
-
[1] 柯美云, 方秀才, 侯晓华. 功能性胃肠病: 肠-脑互动异常[M]. 北京: 科学出版社, 2016: 642-653.
[2] Barberio B, Judge C, Savarino EV, et al. Global prevalence of functional constipation according to the Rome criteria: a systematic review and meta-analysis[J]. Lancet Gastroenterol Hepatol, 2021, 6(8): 638-648. doi: 10.1016/S2468-1253(21)00111-4
[3] Gomes DOVS, Morais MB. Gut microbiota and the use of probiotics in constipation in children and adolescents: systematic review[J]. Rev Paul Pediatr, 2019, 38: e2018123.
[4] 熊杰, 吴雪梅, 庄茜. 结肠传输试验用于功能性便秘患者诊断的临床研究[J]. 海峡药学, 2023, 35(8): 79-82. doi: 10.3969/j.issn.1006-3765.2023.08.021
[5] Yang NN, Ruan MT, Jin SZ. Melanosis coli: a comprehensive review[J]. Gastroenterol Hepatol, 2020, 43(5): 266-272. doi: 10.1016/j.gastrohep.2020.01.002
[6] 柯晓, 刘启鸿, 胡露楠. 功能性便秘中西医诊治研究的思考与策略[J]. 中国中西医结合消化杂志, 2023, 31(11): 828-831. doi: 10.3969/j.issn.1671-038X.2023.11.02
[7] 胡露楠, 刘启鸿, 柯晓, 等. 基于16S rDNA测序技术分析功能性便秘患者粪便菌群[J]. 中国微生态学杂志, 2023, 35(6): 669-673.
[8] 万星阳, 何卓杰, 任东林, 等. 靶向肠道微生态治疗功能性便秘的中西医研究进展[J]. 中国中西医结合消化杂志, 2023, 31(11): 861-867. doi: 10.3969/j.issn.1671-038X.2023.11.08
[9] 廖伟伟, 宁慧娟, 王美娟, 等. 儿童功能性便秘肠道传输功能与肠道微生态的关系[J]. 中国微生态学杂志, 2021, 33(7): 759-764.
[10] 刘启鸿, 方文怡, 胡露楠, 等. 基于16S rRNA高通量测序技术分析福州地区慢传输型便秘患者肠道菌群的变化[J]. 中国微生态学杂志, 2022, 34(3): 323-326.
[11] Matsuura T, Kyokane K, Yamada S, et al. The development of the cure of the functional intestinal disorder based on the differences of gut microbiota in aged patients: a randomized clinical trial[J]. Medicine, 2021, 100(44): e27696. doi: 10.1097/MD.0000000000027696
[12] 胡天骄, 薛鑫, 董秀山. 肠道菌群对肠道运动调节的研究进展[J]. 中国微生态学杂志, 2023, 35(11): 1350-1354.
[13] 李晓宇. 白术内酯Ⅰ调节肠道菌群改善大鼠慢传输型便秘的实验研究[D]. 青岛: 青岛大学, 2022.
[14] Guo MQ, Yao JF, Yang F, et al. The composition of intestinal microbiota and its association with functional constipation of the elderly patients[J]. Future Microbiol, 2020, 15(3): 163-175. doi: 10.2217/fmb-2019-0283
[15] Ross FC, Mayer DE, Horn J, et al. Potential of dietary polyphenols for protection from age-related decline and neurodegeneration: a role for gut microbiota?[J]. Nutr Neurosci, 2024, 27(9): 1058-1076. doi: 10.1080/1028415X.2023.2298098
[16] Hao MQ, Song J, Zhai XH, et al. Improvement of loperamide-hydrochloride-induced intestinal motility disturbance by Platycodon grandiflorum polysaccharides through effects on gut microbes and colonic serotonin[J]. Front Cell Infect Microbiol, 2023, 13: 1105272. doi: 10.3389/fcimb.2023.1105272
[17] Bhattarai Y, Williams BB, Battaglioli EJ, et al. Gut microbiota-produced tryptamine activates an epithelial G-protein-coupled receptor to increase colonic secretion[J]. Cell Host Microbe, 2018, 23(6): 775-785. e5. doi: 10.1016/j.chom.2018.05.004
[18] Shi J, Zhao D, Song SX, et al. High-meat-protein high-fat diet induced dysbiosis of gut microbiota and tryptophan metabolism in wistar rats[J]. J Agric Food Chem, 2020, 68(23): 6333-6346. doi: 10.1021/acs.jafc.0c00245
[19] Rao SS, Seaton K, Miller MJ, et al. Psychological profiles and quality of life differ between patients with dyssynergia and those with slow transit constipation[J]. J Psychosom Res, 2007, 63(4): 441-449. doi: 10.1016/j.jpsychores.2007.05.016
[20] Wang ZH, Liu S, Xu XX, et al. Gut microbiota associated with effectiveness and responsiveness to mindfulness-based cognitive therapy in improving trait anxiety[J]. Front Cell Infect Microbiol, 2022, 12: 719829. doi: 10.3389/fcimb.2022.719829
[21] Zhou YY, Zhang X, Pan LY, et al. Fecal microbiota in pediatric depression and its relation to bowel habits[J]. J Psychiatr Res, 2022, 150: 113-121. doi: 10.1016/j.jpsychires.2022.03.037
-
图(5)
表(1)
计量
- 文章访问数: 942
- PDF下载数: 393
- 施引文献: 0